The simulation of molecular dynamics has always been a challenging task due to the complex interactions between atoms and electrons. However, recent advancements in machine learning algorithms have opened up new possibilities for highly accurate simulations on long time-scales. Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have developed a novel approach that could revolutionize the field of computational chemistry.
The Challenge of Traditional Methods
Traditionally, scientists have relied on solving the Schrödinger equation to compute the interactions of electrons in molecules. This approach is computationally intensive and time-consuming, especially for molecules with a large number of atoms. The need to solve the Schrödinger equation multiple times for molecular dynamics simulations further exacerbates the computational cost, making it a daunting task even on powerful computers.
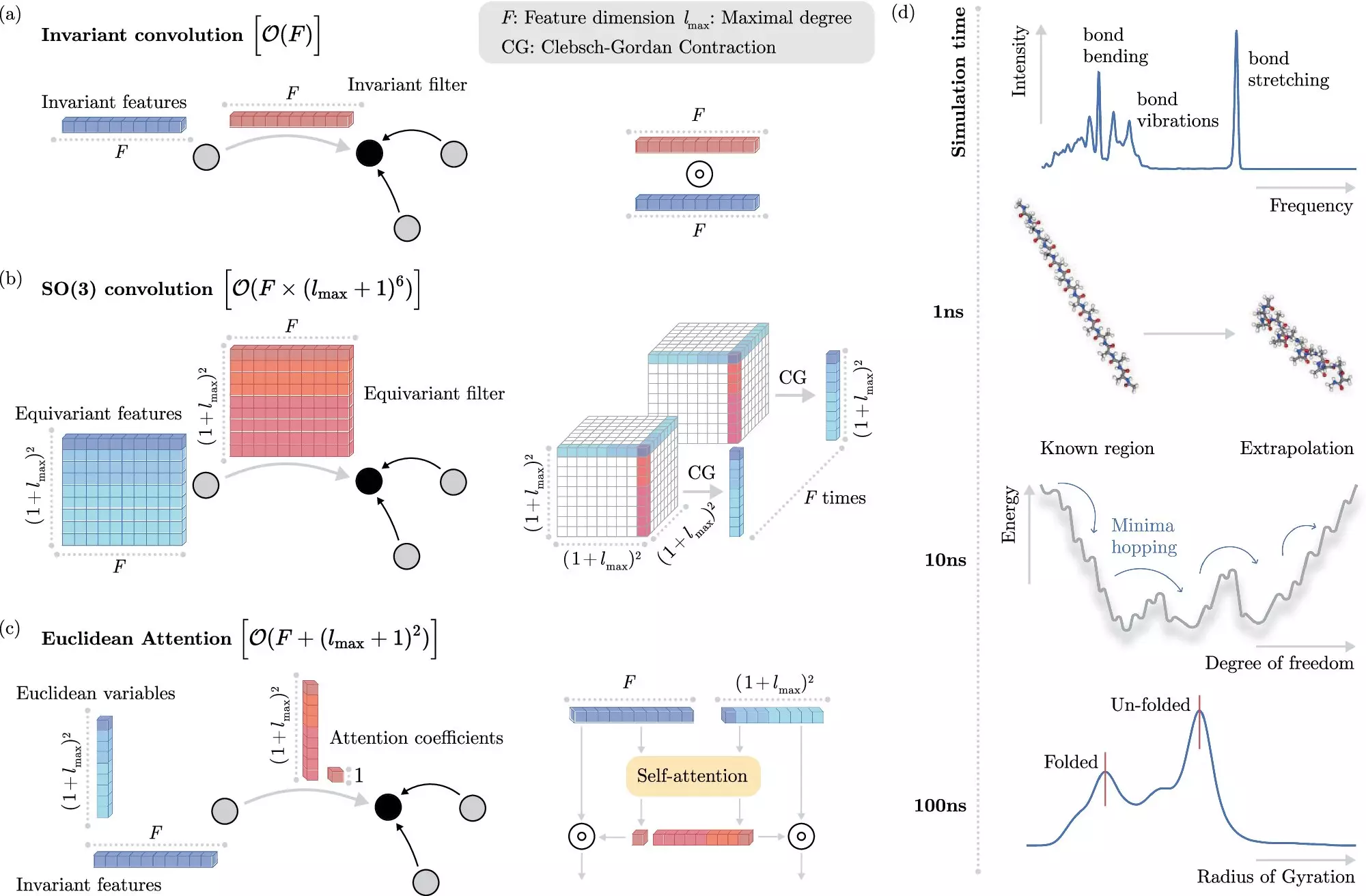
Machine learning methods have emerged as a game-changer in the field of molecular dynamics simulations. Instead of solving the Schrödinger equation explicitly, ML algorithms can learn to predict electronic interactions directly, significantly reducing the computational burden. One key challenge in this approach is teaching the ML system how electrons interact without explicitly modeling them.
The researchers at BIFOLD have developed a groundbreaking learning algorithm that decouples invariances from other information about a chemical system. Unlike previous methods that required extracting invariant components from each operation, this approach simplifies the process and allows the ML model to focus on the most crucial operations. This innovation has drastically reduced the computational cost of simulations, making them more efficient and accessible.
Implications for Research and Development
The new ML algorithm holds tremendous potential for various applications, including drug development and material design. By enabling accurate simulations of molecular interactions, researchers can speed up the process of drug discovery, saving time and resources. The identification of stable molecular structures, such as docosahexaenoic acid, showcases the algorithm’s capabilities in scanning potential candidates with high accuracy, something that was previously infeasible with traditional methods.
As noted by Prof. Dr. Klaus-Robert Müller, the co-director of BIFOLD, the combination of advanced machine learning techniques with physical principles represents a critical advancement in computational chemistry. The ability to scale ML approaches to realistic chemical systems of practical interest is crucial for the future of the field. The next generation of algorithms will need to accurately simulate complex, long-range physical interactions to further advance our understanding of molecular dynamics.
The integration of machine learning with molecular dynamics simulations represents a significant breakthrough in the field of computational chemistry. The novel learning algorithm developed by BIFOLD researchers has the potential to revolutionize how we study and predict the behavior of molecules and materials. By harnessing the power of machine learning, scientists can unravel the mysteries of atomistic systems more efficiently and gain deeper insights into the fundamental processes of nature.
Leave a Reply